Advanced Linux Usage - Variables and Loops
NOTE: in syntax examples, the dollar sign ($) is not to be printed. The dollar sign is usually an indicator that the text following it should be typed in a terminal window.
1. Connecting to UPPMAX
The first step of this lab is to open a ssh connection to UPPMAX. You will need a ssh program to do this:
On Linux: it is included by default, named Terminal.
On OSX: it is included by default, named Terminal.
On Windows: Google MobaXterm and download it.
Fire up the available ssh program and enter the following (replace username with your uppmax user name). -Y means that X-forwarding is activated on the connection, which means graphical data can be transmitted if a program requests it, i.e. programs can use a graphical user interface (GUI) if they want to.
$ ssh -Y username@milou.uppmax.uu.se
and give your password when prompted. As you type, nothing will show on screen. No stars, no dots. It is supposed to be that way. Just type the password and press enter, it will be fine.
Now your screen should look something like this:
2. Getting a node of your own (only if you canceled your job before lunch)
Usually you would do most of the work in this lab directly on one of the login nodes at uppmax, but we have arranged for you to have one core each to avoid disturbances. This was covered briefly in the lecture notes.
Check with squeue -u username if you still have your reservation since before lunch running. If it is running, skip this step and connect to that reservation.(We only have 20 reserved cores, so if someone has two, someone else will not get one..)
# ONLY IF YOU DON'T ALREADY HAVE AN ACTIVE ALLOCATION SINCE BEFORE
$ salloc -A g2016028 -t 04:30:00 -p core -n 1 --no-shell --reservation=g2016028_TUE &
check which node you got (replace username with your uppmax user name)
$ squeue -u username
should look something like this
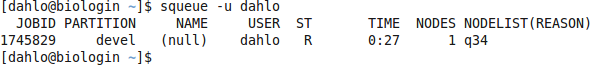
where q34 is the name of the node I got (yours will probably be different). Note the numbers in the Time column. They show for how long the job has been running. When it reaches the time limit you requested (4.5 hours in this case) the session will shut down, and you will lose all unsaved data. Connect to this node from within uppmax.
$ ssh -Y q34
Note: there is a uppmax specific tool called jobinfo that supplies the same kind of information as squeue that you can use as well ($ jobinfo -u username).
3. Copying files needed for this laboratory
To be able to do parts of this lab, you will need some files.
To avoid all the course participants editing the same file all at once, undoing each other’s edits, each participant will get their own copy of the needed files.
The files are located in the folder /sw/courses/ngsintro/loops/
Next, copy the lab files from this folder. -r means recursively, which means all the files including sub-folders of the source folder. Without it, only files directly in the source folder would be copied, NOT sub-folders and files in sub-folders.
NOTE: Remember to tab-complete to avoid typos and too much writing.
Ex.
$ cp -r <source> <destination>
$ cp -r /sw/courses/ngsintro/loops/ ~/glob/ngs-intro/loops
Have a look in ~/glob/ngs-intro/loops:
$ cd ~/glob/ngs-intro/loops
$ ll
If you see files, the copying was successful.
4. Using variables
Variables are like small notes you write stuff on. If you want to save the value of something to be able to use it later, variables is the way to go. Let’s try assigning values to some variables and see how we use them.
$ a=5
Now the values 5 is stored in the variable named a.
To use the variable we have to put a $ sign in front of it so that bash knows we are referring to the variable a and not just typing the letter a.
To see the result of using the variable, we can use the program echo, which prints whatever you give it to the terminal:
$ echo print this text to the terminal
$ echo "you can use quotes if you want to"
As you see, all the words you give it are printed just the way they are. Try putting the variable somewhere in the text:
$ echo Most international flights leave from terminal $a at Arlanda airport
Bash will see that you have a variable there and will replace the variable name with the value the variable have before sending the text to echo.
If you change the value of a and run the exact command again you will see that it changes.
$ a="five"
$ echo Most international flights leave from terminal $a at Arlanda airport
So without changing anything in the echo statement, we can make it output different things, all depending on the value of the variable a.
This is one of the main points of using variables, that you don’t have to change the code or script but you can still make it behave differently depending on the values of the variables.
You can also do mathematics with variables, but we have to tell bash that we want to do calculations first. We do this by wrapping the calculations inside a dollar sign (telling bash it’s a variable) and double parentheses, i.e. $((5+5))
$ a=4
$ echo $a squared is $(($a*$a))
Exercise: Write a echo command that will print out the volume of a rectangular cuboid, with the side lengths specified by variables named x, y, and z. To see that it works correctly, the volume of a rectangular cuboid with sides 5,5,5 is 125, and for 4,5,10 is 200.
Give it a couple of tries on your own first. If you get completely stuck you can see a suggested solution below.
 Click to see how
Click to see how
$ x=4
$ y=5
$ z=10
$ echo The volume of the rectangular cuboid with the sides $x,$y,$z is $(($x*$y*$z)).5. Looping over lists
First off, let’s open another terminal to uppmax so that you have 2 of them open.
Scripting is a lot easier if you have one terminal on the command line ready to run commands and test things, and another one with a text editor where you write the actual code.
That way you will never have to close down the text editor when you want to run the script you are writing on, and then open it up again when you want to continue editing the code.
So open a new terminal window, connect it to uppmax and then connect it to the node you have booked.
Make sure both terminals are in the ~/glob/ngs-intro/loops directory, and start editing a new file with nano where you write your script.
Name the file whatever you want, but in the examples I will refer to it as loop_01.sh.
Write your loops to this file (or create a new file for each new example) and test run it in the other terminal.
The most simple loops are the ones that loop over a predefined list. You saw examples of this in the lecture slides, for example:
for i in "Print these words" one by one;
do
echo $i
done
which will print the value of $i in each iteration of the loop.
Write this loop in the file you are editing with nano, save the file, and then run it in the other terminal you have open.
$ bash loop_01.sh
As you see, the words inside the quotation marks are treated as a single unit, unlike the words after. You can also iterate over numbers, so erase the previous loop you wrote and try this instead:
for number in 1 2 3;
do
echo $number
done
If everything worked correctly you should have gotten the numbers 1 2 3 printed to the screen. As you might guess, this way of writing the list of numbers to iterate over will not be usable once you have more than 10 or so numbers you want to loop over. Fortunately, the creators of bash (and most other computer languages) saw this problem coming a mile away and did something about it. To quickly create a list of numbers in bash, you can use something called a sequence expression to create the list for you.
for whatevernameyouwant in {12..72};
do
echo $whatevernameyouwant
done
Exercise 1
Now that we know how to use variables and create loops, it’s time we start doing something with them.
Let’s say it’s New Year’s Eve and you want to impress your friends with a computerized countdown of the last 10 seconds of the year (haven’t we all?).
Start off with getting a loop to count down from 10 to 0 first.
Notice how fast the computer counts?
That won’t do if it’s seconds we want to be counting down.
Try looking the man page for the sleep command (man sleep) and figure out how to use it.
The point of using sleep is to tell the computer to wait for 1 second after printing the number, instead of rushing to the next iteration in the loop directly.
Try to implement this on your own.
Click to see how
# declare the values the loop will loop over
for secondsToGo in {10..0};
do
# print out the current number
echo $secondsToGo
# sleep for 1 second
sleep 1
done
# declare the start of a new year in a festive manner
echo Happy New Year everyone!!Exercise 2
Let’s try to do something similar to the example in the lecture slides, to run the same commands on multiple files.
In the Uppmax intro we learned how to use samtools to convert bam files to sam files so that humans can read them.
In real life you will never do this, instead you will most likely always do it the other way around.
Sam files take up ~4x more space on the hard drive compared to the same file in bam format, so as soon as you see a sam file you should convert it to a bam file instead to conserve hard drive space.
If you have many sam files that needs converting you don’t want to sit there and type all the commands by hand like some kind of animal.
Write a script that converts all the sam files in a specified directory to bam files.
Incidentally you can find 50 sam files in need of conversion in the folder called sam in the folder you copied to your glob folder earlier in this lab (~/glob/ngs-intro/loops/sam/).
Bonus points if you make the program take the specified directory as an argument, and another bonus point if you get the program to name the resulting bam file to the same name as the sam file but with a .bam ending instead.
Remember that you have to load the samtools module to be able to run it. The way you get samtools to convert a sam file to a bam file is by typing the following command:
samtools view -bS sample_1.sam > sample_1.bam
The -b option tells samtools to output bam format, and the -S option tells samtools that the input is in sam format.
Remember, Google is a good place to get help. If you get stuck, google “bash remove file ending” or “bash argument to script” and look for hits from Stackoverflow/Stackexchange or similar pages. There are always many different way to solve a problem. Try finding one you understand what they do and test if you can get them to work the way you want. If not, look for another solution and try that one instead.
Basic, without bonus points:
Click to see how
# load the modules needed for samtools
module load bioinfo-tools samtools/1.3
# use ls to get the list to iterate over.
# You have to be standing in the correct directory for the script to work
for file in $(ls *.sam);
do
# do the actual converting, just slapping on .bam at the end of the name
samtools view -bS $file > $file.bam
doneAdvanced, with bonus points:
Click to see how
# load the modules needed for samtools
module load bioinfo-tools samtools/1.3
# use ls to get the list to iterate over.
# $1 contains the first argument given to the program
for file in $(ls $1/*.sam);
do
# print a message to the screen so that the user knows what's happening.
# ${file%.*} means that it will take the file name and remove everything
# after the last punctuation in the name.
echo "Converting $file to ${file%.*}.bam"
# do the actual converting
samtools view -bS $file > ${file%.*}.bam
doneExercise 3
Let’s add a small thing to the exercise we just did.
If there already exists a bam file with the same name as the sam file it’s not necessary to convert it again.
Let’s use an if statement to check if the file already exists before we do the conversion.
The following if statement will check if a given filename exists, and prints a message depending on if it exists or not.
FILE=$1
if [ -f $FILE ];
then
echo "File $FILE exists."
else
echo "File $FILE does not exist."
fi
What we want to do is to check if the file doesn’t exists. The way to do that is to invert the answer of the check if the file does exist. To do that in bash, and many other languages, is to use the exclamation mark, which in these kinds of logical situations means “not” or “the opposite of”.
FILE=$1
if [ ! -f $FILE ];
then
echo "File $FILE does not exist."
fi
Now, modify the previous exercise to only do the conversion if a file with the intended name of the bam file doesn’t already exists. I.e. if you have a.sam and want to create a bam file named a.bam, first check if a.bam already exists and only do the conversion if it does not exist.
Basic:
Click to see how
# load the modules needed for samtools
module load bioinfo-tools samtools/1.3
# use ls to get the list to iterate over.
# You have to be standing in the correct directory for the script to work
for file in $(ls *.sam);
do
# check if the intended output file doesn't already exists
if [ ! -f $file.bam ];
then
# do the actual converting, just slapping on .bam at the end of the name
samtools view -bS $file > $file.bam
fi
doneAdvanced:
Click to see how
# load the modules needed for samtools
module load bioinfo-tools samtools/1.3
# use ls to get the list to iterate over.
# $1 contains the first argument given to the program
for file in $(ls $1/*.sam);
do
# check if the intended output file doesn't already exists.
# ${file%.*} means that it will take the file name and remove everything
# after the last punctuation in the name.
if [ ! -f ${file%.*}.bam ];
then
# print a message to the screen so that the user knows what's happening.
echo "Converting $file to ${file%.*}.bam"
# do the actual converting
samtools view -bS $file > ${file%.*}.bam
else
# inform the user that the conversion is skipped
echo "Skipping conversion of $file as ${file%.*}.bam already exist"
fi
doneBonus exercise 1
Maths and programming are usually a very good combination, so many of the examples of programming you’ll see involve some kind of maths.
Now we will write a loop that will calculate the factorial of a number.
As wikipedia will tell you, “the factorial of a non-negative integer n, denoted by n!, is the product of all positive integers less than or equal to n”, i.e. multiply all the integers, starting from 1, leading up to and including a number with each other.
The factorial of 5, written 5!, would be 1 * 2 * 3 * 4 * 5 = 120
Doing this by hand would start taking its time even after a couple of steps, but since we know how to loop that should not be a problem anymore.
Write a loop that will calculate the factorial of a given number stored in the variable $n.
A problem you will encounter is that the sequence expression, {1..10}, from before doesn’t handle variables. This is because of the way bash is built. The sequence expressions are handled before handling the variables so when bash tries to generate the sequence, the variable names have not yet been replaced with the values they contain. This leads to bash trying to create a sequence from 1 to $n, which of course doesn’t mean anything.
To get around this we can use a different way of generating sequences (there are always alternatives).
There is a program called seq that does pretty much the same thing as the sequence expression, and since it is a program it will be executed after the variables have been handled.
It’s as easy to use as the sequence expressions; instead of writing {1..10} just write $( seq 1 10 ).
Click to see how
# set the number you want to calculate the factorial of
n=10
# you have to initialize a variable before you can start using it.
# Leaving this empty would lead to the first iteration of the loop trying
# to use a variable that has no value, which would cause it to crash
factorial=1
# declare the values the loop will loop over (1 to whatever $n is)
for i in $( seq 1 $n );
do
# set factorial to whatever factorial is at the moment, multiplied with the variable $i
factorial=$(( $factorial * $i ))
done
# print the result
echo The factorial of $n is $factorialBonus exercise 2
Now, let’s combine everything you’ve learned so far in this course.
Write a script that runs the pipeline from the file types exercise for each fastq file in a specified directory, using the same reference genome as in the file type exercise.
If that sounds too easy, make the script submit a slurm job for each sample that will run the pipeline for that sample on a calculation node (1 core, 5 minutes each).
And if that is too easy, add that the pipeline will use the local hard drive on the calculation node for all files used in the analysis.
When the analysis is done, only fastq files and sorted and indexed bam files should be in your glob folder.
Read more about the $SNIC_TMP variable in the disk storage guide on the UPPMAX homepage.
There is a bunch of fastq files in the directory ~/glob/ngs-intro/loops/fastq/ that is to be used for this exercise.
Basic solution:
Click to see how
# make the dummy pipeline available
export PATH=$PATH:/sw/courses/ngsintro/uppmax_pipeline_exercise/dummy_scripts
# index the reference genome
reference_indexer -r ~/glob/ngs-intro/filetypes/0_ref/ad2.fa
# go to the input files
cd $1
# loop over all the fastq files
for file in $(ls *.fastq);
do
# save the file name without the path information for convenience
file_basename=$(basename $file)
# align the reads
align_reads -r ~/glob/ngs-intro/filetypes/0_ref/ad2.fa -i $file_basename -o $file_basename.sam
# convert the sam file to a bam file
sambam_tool -f bam -i $file_basename.sam -o $file_basename.bam
# sort the bam file
sambam_tool -f sort -i $file_basename.bam -o $file_basename.sorted.bam
# index the bam file
sambam_tool -f index -i $file_basename.sorted.bam
doneAdvanced solution:
Click to see how
# make the dummy pipeline available in this script
export PATH=$PATH:/sw/courses/ngsintro/uppmax_pipeline_exercise/dummy_scripts
# index the reference genome once, only if needed
if [ ! -f ~/glob/ngs-intro/filetypes/0_ref/ad2.fa.idx ];
then
reference_indexer -r ~/glob/ngs-intro/filetypes/0_ref/ad2.fa
fi
# find out the absolute path to the input files
cd $1
input_absolute_path=$(pwd)
# go back to the previous directory now that the absolute path has been saved
cd -
# loop over all the fastq files
for file in $(ls $input_absolute_path/*.fastq);
do
# print status report
echo Processing $file
# save the file name without the path information for convenience
file_basename=$(basename $file)
# save the file name without the file ending for convenience
file_prefix=${file_basename%.*}
# print a temporary script file that will be submitted to slurm
echo "#!/bin/bash -l
#SBATCH -A g2016028
#SBATCH -p core
#SBATCH -n 1
#SBATCH -t 00:05:00
#SBATCH -J $file_basename
# make the dummy pipeline available on the calculation node
echo "Loading modules"
export PATH=\$PATH:/sw/courses/ngsintro/uppmax_pipeline_exercise/dummy_scripts
# copy the reference genome, index and sample file to the nodes local hard drive.
# You have to escape the dollar sign in SNIC_TMP to keep bash from resolving
# it to it's value in the submitter script already.
echo "Copying data to node local hard drive"
cp ~/glob/ngs-intro/filetypes/0_ref/ad2.fa* $file \$SNIC_TMP/
# go the the nodes local hard drive
echo "Changing directory to node local hard drive"
cd \$SNIC_TMP
# align the reads
echo "Aligning the reads"
align_reads -r ad2.fa -i $file_basename -o $file_prefix.sam
# convert the sam file to a bam file
echo "Converting sam to bam"
sambam_tool -f bam -i $file_prefix.sam -o $file_prefix.bam
# sort the bam file
echo "Sorting the bam file"
sambam_tool -f sort -i $file_prefix.bam -o $file_prefix.sorted.bam
# index the bam file
echo "Indexing the sorted bam file"
sambam_tool -f index -i $file_prefix.sorted.bam
# copy back the files you want to keep
echo "Copying results back to network storage"
cp $file_prefix.sorted.bam $input_absolute_path/
cp $file_prefix.sorted.bam.bai $input_absolute_path/$file_prefix.sorted.bai
echo "Finished"
" > tmp.sbatch
# submit the temporary script file
sbatch tmp.sbatch
done
# remove the temporary file now that everything has been submitted
rm tmp.sbatch