2 Main exercise
The main exercise covers Differential Gene Expression (DGE) workflow from raw reads to a list of differentially expressed genes.
2.1 Preparation
For Linux and Mac users, Log in to Uppmax in a way so that the generated graphics are exported via the network to your screen. Login in to Uppmax with X-forwarding enabled. This will allow any graphical interface that you start on your compute node to be exported to your computer.
Linux users are recommended to use this:
ssh -X username@rackham.uppmax.uu.seAnd Mac user are recommended to use this:
ssh -Y username@rackham.uppmax.uu.seWindows users on MobaXterm do not need to worry about this option.
2.1.1 Book a node
For the RNA-Seq part of the course (Thu/Fri), we will work on the Snowy cluster. A standard compute node on cluster Snowy has 128 GB of RAM and 16 cores. Therefore, each core gives you 8 GB of RAM. We will use 8 cores per person for this session which gives you about 64 GB RAM. The code below is valid to run at the start of the day. If you are running it in the middle of a day, you need to decrease the time (-t). Do not run this twice and also make sure you are not running computations on a login node.
To run jobs on the snowy cluster therefore, we need to add -M snowy.
Book resources for RNA-Seq day 1.
salloc -A snic2019-8-3 -t 08:00:00 -p core -n 8 --reservation=snic2019-8-3_7 -M snowyBook resources for RNA-Seq day 2.
salloc -A snic2019-8-3 -t 08:00:00 -p core -n 8 --reservation=snic2019-8-3_8 -M snowy2.1.2 Set-up directory
Setting up the directory structure is an important step as it helps to keep our raw data, intermediate data and results in an organised manner. All work must be carried out at this location /proj/snic2019-8-3/nobackup/[user]/ where [user] is your user name. All RNA-Seq related activities must be carried out in a sub-directory named rnaseq.
Set up the below directory structure in your project directory.
[user]/
rnaseq/
+-- 1_raw/
+-- 2_fastqc/
+-- 3_mapping/
+-- 4_qualimap/
+-- 5_dge/
+-- 6_multiqc/
+-- reference/
| +-- mouse/
| +-- mouse_chr11/
+-- scripts/mkdir rnaseq cd rnaseq mkdir 1_raw 2_fastqc 3_mapping 4_qualimap 5_dge 6_multiqc reference scripts cd reference mkdir mouse mkdir mouse_chr11 cd ..
The 1_raw directory will hold the raw fastq files (soft-links). 2_fastqc will hold FastQC outputs. 3_mapping will hold the STAR mapping output files. 4_qualimap will hold the QualiMap output files. 5_dge will hold the counts from featureCounts and all differential gene expression related files. 6_multiqc will hold MultiQC outputs. reference directory will hold the reference genome, annotations and STAR indices.
It might be a good idea to open an additional terminal window. One to navigate through directories and another for scripting in the scripts directory.
2.1.3 Create symbolic links
We have the raw fastq files in this remote directory: /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/. We are going to create symbolic links (soft-links) for these files from our 1_raw directory to the remote directory. We do this because they are large files and simply copying them would use up a lot of storage space. Soft-linking files and folders allows us to work with those files as if they were actually there. Use pwd to check if you are standing in the correct directory. You should be standing here:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/1_rawRun below to create softlinks.
ln -s /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/*.fastq.gz .Check if your files have linked correctly. You should be able to see as below.
[user@rackham2 1_raw]$ ls -l
SRR3222409_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222409_1.fastq.gz
SRR3222409_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222409_2.fastq.gz
SRR3222410_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222410_1.fastq.gz
SRR3222410_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222410_2.fastq.gz
SRR3222411_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222411_1.fastq.gz
SRR3222411_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222411_2.fastq.gz
SRR3222412_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222412_1.fastq.gz
SRR3222412_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222412_2.fastq.gz
SRR3222413_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222413_1.fastq.gz
SRR3222413_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222413_2.fastq.gz
SRR3222414_1.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222414_1.fastq.gz
SRR3222414_2.fastq.gz -> /sw/share/compstore/courses/ngsintro/rnaseq/main/1_raw/SRR3222414_2.fastq.gz2.2 FastQC: Quality check
After receiving raw reads from a high throughput sequencing centre it is essential to check their quality. FastQC provides a simple way to do some quality control check on raw sequence data. It provides a modular set of analyses which you can use to get a quick impression of whether your data has any problems of which you should be aware before doing any further analysis.
Change into the 2_fastqc directory. Use pwd to check if you are standing in the correct directory. You should be standing here:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/2_fastqcLoad Uppmax modules bioinfo-tools and FastQC FastQC/0.11.5.
module load bioinfo-tools
module load FastQC/0.11.5Once the module is loaded, FastQC program is available through the command fastqc. Use fastqc --help to see the various parameters available to the program. We will use -t 8, to specify number of threads, -o to specify the output directory path and finally, the name of the input fastq file to analyse. The syntax will look like below.
fastqc -t 8 -o . ../1_raw/filename.fastq.gzBased on the above command, we will write a bash loop to process all fastq files in the directory. Writing multi-line commands through the terminal can be a pain. Therefore, we will run larger scripts from a bash script file. Move to your scripts directory and create a new file named fastqc.sh.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/scriptstouch fastqc.sh
Use nano,vim or gedit to edit fastqc.sh.
#!/bin/bash
for i in ../1_raw/*.fastq.gz
do
echo "Running $i ..."
fastqc -t 8 -o . "$i"
done
While standing in the 2_fastqc directory, run the file fastqc.sh. Use pwd to check if you are standing in the correct directory.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/2_fastqcbash ../scripts/fastqc.sh
After the fastqc run, there should be a .zip file and a .html file for every fastq file. The .html file is the report that you need. Open the .html in the browser and view it. You only need to do this for one file now. We will do a comparison with all samples when using the MultiQC tool.
firefox file.html & Adding & at the end sends that process to the background, so that the console is free to accept new commands.
Optional
Download the .html file to your computer and view it.
All users can use an SFTP browser like Filezilla or Cyberduck for a GUI interface. Windows users can also use the MobaXterm SFTP file browser to drag and drop. Linux and Mac users can use SFTP or SCP by running the below command in a LOCAL terminal and NOT on Uppmax.
scp user@rackham.uppmax.uu.se:/proj/snic2019-8-3/nobackup/[user]/2_fastqc/SRR3222409_1_fastqc.html ./Go back to the FastQC website and compare your report with the sample report for Good Illumina data and Bad Illumina data.
Discuss based on your reports, whether your data is of good enough quality and/or what steps are needed to fix it.
2.3 STAR: Mapping
After verifying that the quality of the raw sequencing reads is acceptable, we will map the reads to the reference genome. There are many mappers/aligners available, so it may be good to choose one that is adequate for your type of data. Here, we will use a software called STAR (Spliced Transcripts Alignment to a Reference) as it is good for generic purposes, fast, easy to use and has been shown to outperform many of the other tools when aligning 2x76bp paired-end data. Before we begin mapping, we need to obtain genome reference sequence (.fasta file) and a corresponding annotation file (.gtf) and build a STAR index. Due to time constraints, we will practice index building only on chromosome 11. But, then we will use the pre-prepared full-genome index to run the actual mapping.
2.3.1 Get reference
It is best if the reference genome (.fasta) and annotation (.gtf) files come from the same source to avoid potential naming conventions problems. It is also good to check in the manual of the aligner you use for hints on what type of files are needed to do the mapping.
What is the idea behind building STAR index? What files are needed to build one? Where do we take them from? Could one use a STAR index that was generated before? Browse through Ensembl and try to find the files needed. Note that we are working with Mouse (Mus musculus).
Move into the reference directory and download the Chr 11 genome (.fasta) file and the genome-wide annotation file (.gtf) from Ensembl.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/referencewget ftp://ftp.ensembl.org/pub/release-93/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.chromosome.11.fa.gz wget ftp://ftp.ensembl.org/pub/release-93/gtf/mus_musculus/Mus_musculus.GRCm38.93.gtf.gz
Decompress the files for use.
+gunzip Mus_musculus.GRCm38.dna.chromosome.11.fa.gz gunzip Mus_musculus.GRCm38.93.gtf.gz
You should now have the files as below.
[user@rackham2 reference]$ ll
drwxrwsr-x 2 user g201XXXX 4.0K Sep 4 19:33 mouse
drwxrwsr-x 2 user g201XXXX 4.0K Sep 4 19:32 mouse_chr11
-rw-rw-r-- 1 user g201XXXX 742M Sep 4 19:31 Mus_musculus.GRCm38.93.gtf
-rw-rw-r-- 1 user g201XXXX 119M Sep 4 19:31 Mus_musculus.GRCm38.dna.chromosome.11.fa2.3.2 Build index
Move into the reference directory if not already there. Load module STAR version 2.5.2b. Remember to load bioinfo-tools if you haven’t done so already.
module load bioinfo-tools
module load star/2.5.2b To search for other available versions of STAR, use module spider star.
Create a new bash script in your scripts directory named star_index.sh and add the following lines:
#!/bin/bash
# load module
module load bioinfo-tools
module load star/2.5.2b
star \
--runMode genomeGenerate \
--runThreadN 8 \
--genomeDir ./mouse_chr11 \
--genomeFastaFiles ./Mus_musculus.GRCm38.dna.chromosome.11.fa \
--sjdbGTFfile ./Mus_musculus.GRCm38.93.gtfThe above script means that STAR should run in genomeGenerate mode to build an index. It should use 8 threads for computation. The output files must be directed to the indicated directory. The paths to the .fasta file and the annotation file (.gtf) is also shown.
Run the script from the reference directory. Use pwd to check if you are standing in the correct directory.
bash ../scripts/star_index.shOnce the indexing is complete, move into the mouse_chr11 directory and make sure you have all the files.
[user@rackham2 mouse_chr11]$ ll
-rw-rw-r-- 1 user g201XXXX 10 Sep 4 19:31 chrLength.txt
-rw-rw-r-- 1 user g201XXXX 13 Sep 4 19:31 chrNameLength.txt
-rw-rw-r-- 1 user g201XXXX 3 Sep 4 19:31 chrName.txt
-rw-rw-r-- 1 user g201XXXX 12 Sep 4 19:31 chrStart.txt
-rw-rw-r-- 1 user g201XXXX 1.7M Sep 4 19:33 exonGeTrInfo.tab
-rw-rw-r-- 1 user g201XXXX 805K Sep 4 19:33 exonInfo.tab
-rw-rw-r-- 1 user g201XXXX 56K Sep 4 19:33 geneInfo.tab
-rw-rw-r-- 1 user g201XXXX 121M Sep 4 19:33 Genome
-rw-rw-r-- 1 user g201XXXX 553 Sep 4 19:31 genomeParameters.txt
-rw-rw-r-- 1 user g201XXXX 967M Sep 4 19:33 SA
-rw-rw-r-- 1 user g201XXXX 1.5G Sep 4 19:33 SAindex
-rw-rw-r-- 1 user g201XXXX 522K Sep 4 19:33 sjdbInfo.txt
-rw-rw-r-- 1 user g201XXXX 463K Sep 4 19:33 sjdbList.fromGTF.out.tab
-rw-rw-r-- 1 user g201XXXX 463K Sep 4 19:33 sjdbList.out.tab
-rw-rw-r-- 1 user g201XXXX 480K Sep 4 19:33 transcriptInfo.tabThis index for chr11 was created just to familiarise with the steps. We will use the index built on the whole genome for downstream exercises. The index for the whole genome was prepared for us before class in the very same way as for the chromosome 11 in steps above. It just requires more time (ca. 4h) to run. The index is found here: /sw/share/compstore/courses/ngsintro/rnaseq/reference/mouse/.
Soft-link all the files inside /sw/share/compstore/courses/ngsintro/rnaseq/reference/mouse/ to the directory named mouse which is inside your rnaseq/reference/.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/referencecd mouse ln -s /sw/share/compstore/courses/ngsintro/rnaseq/reference/mouse/* .
2.3.3 Map reads
Now that we have the index ready, we are ready to map reads. Move to the directory 3_mapping. Use pwd to check if you are standing in the correct directory.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/3_mappingWe will create softlinks to the fastq files from here to make things easier.
+cd 3_mapping ln -s ../1_raw/* .
These are the parameters that we want to specify for the STAR mapping run:
- Run mode is now
alignReads - Specify the full genome index path
- Specify the number of threads
- We must indicate the input is gzipped and must be uncompressed
- Indicate read1 and read2 since we have paired-end reads
- Specify the annotation (.gtf) file
- Specify an output file name
- Specify that the output must be BAM and the reads must be sorted by coordinate
Our mapping script will look like this:
+star \ --runMode alignReads \ --genomeDir "../reference/mouse" \ --runThreadN 8 \ --readFilesCommand zcat \ --readFilesIn sample_1.fastq.gz sample_2.fastq.gz \ --sjdbGTFfile "../reference/Mus_musculus.GRCm38.93.gtf" \ --outFileNamePrefix "sample1" \ --outSAMtype BAM SortedByCoordinate
But, we will generalise the above script to be used as a bash script to read any two input files and to automatically create the output filename.
Now create a new bash script file named star_align.sh in your scripts directory and add the script below to it.
#!/bin/bash
module load bioinfo-tools
module load star/2.5.2b
# create output file name
prefix="${1/_*/}"
star \
--runMode alignReads \
--genomeDir "../reference/mouse" \
--runThreadN 8 \
--readFilesCommand zcat \
--readFilesIn $1 $2 \
--sjdbGTFfile "../reference/Mus_musculus.GRCm38.93.gtf" \
--outFileNamePrefix "$prefix" \
--outSAMtype BAM SortedByCoordinate
In the above script, the two input fastq files as passed in as parameters $1 and $2. The output filename is automatically created using this line prefix='${1/_*/}' from input filename of $1. For example, a file named sample_1.fastq.gz will have the _1.fastq.gz removed and the prefix will be just sample. This approach will work only if your filenames are labelled suitably.
Now we can run the bash script like below while standing in the 3_mapping directory.
bash ../scripts/star_align.sh sample_1.fastq.gz sample_2.fastq.gzNow, do the same for the other samples as well if you have time. Otherwise just run for one sample and results for the other samples can be copied (See end of this section).
Optional
Try to create a new bash loop script (star_align_batch.sh) to iterate over all fastq files in the directory and run the mapping using the star_align.sh script. Note that there is a bit of a tricky issue here. You need to use two fastq files (_1 and _2) per run rather than one file.
## find only files for read 1 and extract the sample name
lines=$(find *_1.fastq.gz | sed "s/_1.fastq.gz//")
for i in ${lines}
do
## use the sample name and add suffix (_1.fastq.gz or _2.fastq.gz)
echo "Mapping ${i}_1.fastq.gz and ${i}_2.fastq.gz ..."
bash ../scripts/star_align.sh "${i}_1.fastq.gz ${i}_2.fastq.gz"
done
Run the star_align_batch.sh script in the 3_mapping directory.
bash ../scripts/star_align_batch.sh
At the end of the mapping jobs, you should have the following list of output files for every sample:
[user@rackham2 3_mapping]$ ls -l
-rw-rw-r-- 1 user g201XXXX 628M Sep 6 00:54 SRR3222409Aligned.sortedByCoord.out.bam
-rw-rw-r-- 1 user g201XXXX 1.9K Sep 6 00:54 SRR3222409Log.final.out
-rw-rw-r-- 1 user g201XXXX 21K Sep 6 00:54 SRR3222409Log.out
-rw-rw-r-- 1 user g201XXXX 482 Sep 6 00:54 SRR3222409Log.progress.out
-rw-rw-r-- 1 user g201XXXX 3.6M Sep 6 00:54 SRR3222409SJ.out.tab
drwx--S--- 2 user g201XXXX 4.0K Sep 6 00:50 SRR3222409_STARgenomeThe .bam file contains the alignment of all reads to the reference genome in binary format. BAM files are not human readable directly. To view a BAM file in text format, you can use samtools view functionality.
module load samtools/1.6
samtools view SRR3222409Aligned.sortedByCoord.out.bam | head
SRR3222409.8816556 163 1 3199842 255 101M = 3199859 116 TTTTAAAGTTTTACAAGAAAAAAAATCAGATAACCGAGGAAAATTATTCATTATGAAGTACTACTTTCCACTTCATTTCATCACAAATTGTAACTTACTTA DDBDDIIIHIIHHHIHIHHIIIIIDHHIIIIIIIIIIIIIIHIIIIHIIIEHHIIIHIIIIGIIIIIIIIIIIIIIHIIHEHIIIIIIHIIIIIHIIIIII NH:i:1 HI:i:1 AS:i:198 nM:i:0
SRR3222409.8816556 83 1 3199859 255 99M = 3199842 -116AAAAAAAATCAGATAACCGAGGAAAATTATTCATTATGAAGTACTACTTTCCACTTCATTTCATCACAAATTGTAACTTACTTAACTGACCAAAAAAAC IIIIIHHIHHIIIIHHEEHIIIHIIHHHIHIIIIIIIHIHHIIIIIIHIIIIIIIIHHHHHIIIIIHIHHIIIHIHHFHHIIHIIIIHCIIIIHDDD@D NH:i:1 HI:i:1 AS:i:198 nM:i:0
SRR3222409.2149741 163 1 3199933 255 101M = 3200069 237 AACTTACTTAACTGACCAAAAAAACTATGGTACTGCAGTATAGCAAATACTCCACACACTGTGCTTTGAGCTAGAGCACTTGGAGTCACTGCCCAGGGCAG ABDDDHHIIIIIIIIIIIIIIIHHIIIIIIIIIIIIIIIIIIIIIIII<<FHIHGHIIIIGIHEHIIIIIGIIIIIIIIIIIIIIHIIIIIHIIIIHIIIH NH:i:1 HI:i:1 AS:i:200 nM:i:0Can you identify what some of these columns are?
The Log.final.out file gives a summary of the mapping run. This file is used by MultiQC later to collect mapping statistics.
Inspect one of the mapping log files to identify the number of uniquely mapped reads and multi-mapped reads.
+cat SRR3222409Log.final.out
Started job on | Sep 08 14:03:46
Started mapping on | Sep 08 14:07:01
Finished on | Sep 08 14:09:05
Mapping speed, Million of reads per hour | 154.78
Number of input reads | 5331353
Average input read length | 201
UNIQUE READS:
Uniquely mapped reads number | 4532497
Uniquely mapped reads % | 85.02%
Average mapped length | 199.72
Number of splices: Total | 2628072
Number of splices: Annotated (sjdb) | 2608823
Number of splices: GT/AG | 2604679
Number of splices: GC/AG | 15762
Number of splices: AT/AC | 2422
Number of splices: Non-canonical | 5209
Mismatch rate per base, % | 0.18%
Deletion rate per base | 0.02%
Deletion average length | 1.49
Insertion rate per base | 0.01%
Insertion average length | 1.37
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 493795
% of reads mapped to multiple loci | 9.26%
Number of reads mapped to too many loci | 8241
% of reads mapped to too many loci | 0.15%
UNMAPPED READS:
% of reads unmapped: too many mismatches | 0.00%
% of reads unmapped: too short | 5.51%
% of reads unmapped: other | 0.06%
CHIMERIC READS:
Number of chimeric reads | 0
% of chimeric reads | 0.00%
The BAM file names can be simplified by renaming them. This command renames all BAM files.
rename "Aligned.sortedByCoord.out" "" *.bamNext, we need to index these BAM files. Indexing creates .bam.bai files which are required by many downstream programs to quickly and efficiently locate reads anywhere in the BAM file.
Index all BAM files.
module load samtools/1.8
for i in *.bam
do
echo "Indexing $i ..."
samtools index $i
doneFinally, we should have .bai index files for all BAM files.
[user@rackham2 3_mapping]$ ls -l
-rw-rw-r-- 1 user g201XXXX 628M Sep 6 00:54 SRR3222409.bam
-rw-rw-r-- 1 user g201XXXX 1.8M Sep 6 01:22 SRR3222409.bam.bai If you are running short of time or unable to run the mapping, you can copy over results for all samples that have been prepared for you before class. They are available at this location: /sw/share/compstore/courses/ngsintro/rnaseq/main/3_mapping/.
cp -r /sw/share/compstore/courses/ngsintro/rnaseq/main/3_mapping/* /proj/snic2019-8-3/nobackup/[user]/rnaseq/3_mapping/2.4 QualiMap: Post-alignment QC
Some important quality aspects, such as saturation of sequencing depth, read distribution between different genomic features or coverage uniformity along transcripts, can be measured only after mapping reads to the reference genome. One of the tools to perform this post-alignment quality control is QualiMap. QualiMap examines sequencing alignment data in SAM/BAM files according to the features of the mapped reads and provides an overall view of the data that helps to the detect biases in the sequencing and/or mapping of the data and eases decision-making for further analysis.
Read through QualiMap documentation and see if you can figure it out how to run it to assess post-alignment quality on the RNA-seq mapped samples. Here is the RNA-Seq specific tool explanation. The tool is already installed on Uppmax as a module.
Load the QualiMap module version 2.2.1 and create a bash script named qualimap.sh in your scripts directory.
Add the following script to it.
+#!/bin/bash
# load modules
module load bioinfo-tools
module load QualiMap/2.2.1
prefix="${1/.bam/}"
export DISPLAY=""
qualimap rnaseq -pe \
-bam $1 \
-gtf "../reference/Mus_musculus.GRCm38.93.gtf" \
-outdir "$prefix" \
-outfile "$prefix" \
-outformat "HTML" \
--java-mem-size=50G >& "${prefix}-qualimap.log"
The line prefix="${1/.bam/}" is used to remove .bam from the input filename and create a prefix which will be used to label output. The export DISPLAY="" is used to run JAVA application in headless mode or else throws an error about X11 display. The last part >& "${prefix}-qualimap.log" saves the standard-out as a log file.
create a new bash loop script named qualimap_batch.sh with a bash loop to run the qualimap script over all BAM files. The loop should look like below. Alternatively, you can also simply run the script below directly on the command line.
for i in ../3_mapping/*.bam
do
echo "Running QualiMap on $i ..."
bash ../scripts/qualimap.sh $i
done
Run the loop script qualimap_batch.sh in the directory 4_qualimap.
bash ../scripts/qualimap_batch.sh
Qualimap should have created a directory for every BAM file. Inside every directory, you should see:
[user@rackham2 4_qualimap]$ ls -l
drwxrwxr-x 2 user g201XXXX 4.0K Sep 14 17:24 css
drwxrwxr-x 2 user g201XXXX 4.0K Sep 14 17:24 images_qualimapReport
-rw-rw-r-- 1 user g201XXXX 11K Sep 14 17:24 qualimapReport.html
drwxrwxr-x 2 user g201XXXX 4.0K Sep 14 17:24 raw_data_qualimapReport
-rw-rw-r-- 1 user g201XXXX 1.2K Sep 14 17:24 rnaseq_qc_results.txtInspect the HTML output file and try to make sense of it.
+firefox qualimapReport.html &
If you are running out of time or were unable to run QualiMap, you can also copy pre-run QualiMap output from this location: /sw/share/compstore/courses/ngsintro/rnaseq/main/4_qualimap/.
cp -r /sw/share/compstore/courses/ngsintro/rnaseq/main/4_qualimap/* /proj/snic2019-8-3/nobackup/[user]/rnaseq/4_qualimap/Check the QualiMap report for one sample and discuss if the sample is of good quality. You only need to do this for one file now. We will do a comparison with all samples when using the MultiQC tool.
2.5 featureCounts: Counting reads
After ensuring mapping quality, we can move on to enumerating reads mapping to genomic features of interest. Here we will use featureCounts, an ultrafast and accurate read summarization program, that can count mapped reads for genomic features such as genes, exons, promoter, gene bodies, genomic bins and chromosomal locations.
Read featureCounts documentation and see if you can figure it out how to use paired-end reads using an unstranded library to count fragments overlapping with exonic regions and summarise over genes.
Load the subread module version 1.5.2 on Uppmax. Create a bash script named featurecounts.sh in the directory scripts.
We could run featureCounts on each BAM file, produce a text output for each sample and combine the output. But the easier way is to provide a list of all BAM files and featureCounts will combine counts for all samples into one text file.
Below is the script that we will use:
+#!/bin/bash # load modules module load bioinfo-tools module load subread/1.5.2 featureCounts \ -a "../reference/Mus_musculus.GRCm38.93.gtf" \ -o "counts.txt" \ -F "GTF" \ -t "exon" \ -g "gene_id" \ -p \ -s 0 \ -T 8 \ ../3_mapping/*.bam
In the above script, we indicate the path of the annotation file (-a "../reference/Mus_musculus.GRCm38.93.gtf"), specify the output file name (-o "counts.txt"), specify that that annotation file is in GTF format (-F "GTF"), specify that reads are to be counted over exonic features (-t "exon") and summarised to the gene level (-g "gene_id"). We also specify that the reads are paired-end (-p), the library is unstranded (-s 0) and the number of threads to use (-T 8).
Run the featurecounts bash script in the directory 5_dge. Use pwd to check if you are standing in the correct directory.
You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/5_dgebash ../scripts/featurecounts.sh
You should have two files:
[user@rackham2 5_dge]$ ls -l
-rw-rw-r-- 1 user g201XXXX 2.8M Sep 15 11:05 counts.txt
-rw-rw-r-- 1 user g201XXXX 658 Sep 15 11:05 counts.txt.summaryInspect the files and try to make sense of them.
2.6 MultiQC: Combined QC report
We will use the tool MultiQC to crawl through the output, log files etc from FastQC, STAR, QualiMap and featureCounts to create a combined QC report.
Run MultiQC as shown below in the 6_multiqc directory. You should be standing here to run this:
/proj/snic2019-8-3/nobackup/[user]/rnaseq/6_multiqcmodule load bioinfo-tools
module load MultiQC/1.6
multiqc --interactive ../You should have two files:
[user@rackham2 6_multiqc]$ ls -l
drwxrwsr-x 2 user g201XXXX 4.0K Sep 6 22:33 multiqc_data
-rw-rw-r-- 1 user g201XXXX 1.3M Sep 6 22:33 multiqc_report.html Open the MultiQC HTML report using firefox and/or transfer to your computer and inspect the report.
firefox multiqc_report.html &
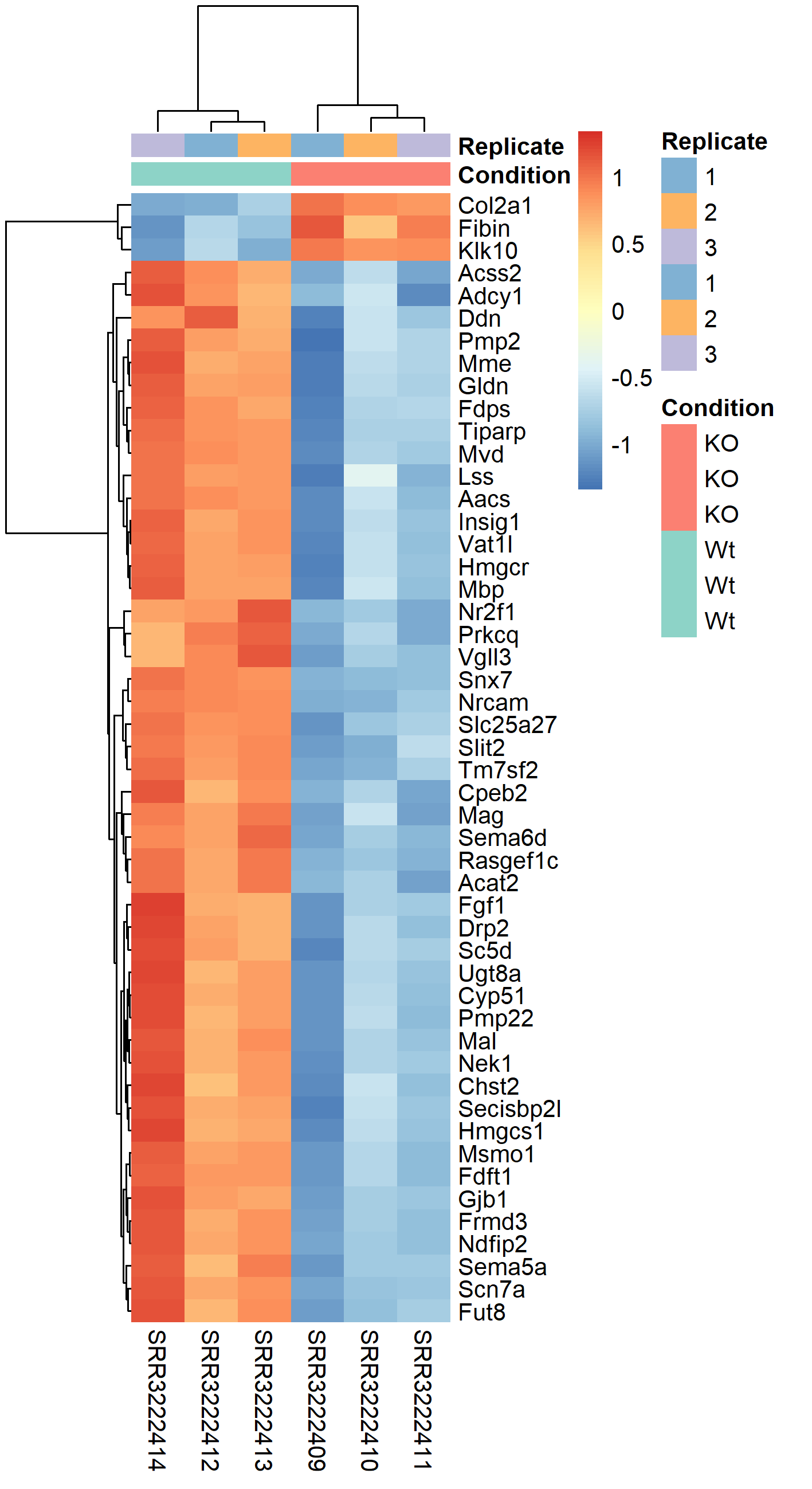
2.7 Differential gene expression
The easiest way to perform differential expression is to use one of the statistical packages, within R environment, that were specifically designed for analyses of read counts arising from RNA-seq, SAGE and similar technologies. Here, we will one of such packages called edgeR. Learning R is beyond the scope of this course so we prepared basic ready to run R scripts to find DE genes between conditions KO and Wt.
Move to the 5_dge directory and load R modules for use.
module load R/3.4.3
module load R_packages/3.4.3Use pwd to check if you are standing in the correct directory. Copy the following files to the 5_dge directory.
/sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/annotations.txt/sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/dge.R
Make sure you have the counts.txt file from featureCounts. If not, you can copy this file too.
/sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/counts.txt
cp /sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/annotations.txt .
cp /sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/dge.R .
cp /sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/counts.txt .Now, run the R script in 5_dge directory.
Rscript dge.RThis should have produced the following output files:
[user@rackham2 5_dge]$ ls -l
-rw-rw-r-- 1 user g201XXXX 8.9M Nov 29 16:31 dge_data.RData
-rw-rw-r-- 1 user g201XXXX 2.6M Nov 29 16:31 dge_results.txt Copy the results text file (dge_results.txt) to your computer and inspect the results. What are the columns? How many differentially expressed genes are present at an FDR cutoff of 0.05? How many genes are upregulated and how many are down-regulated? How does this change if we set a fold-change cut-off of 1?
Open in a spreadsheet editor like Microsoft Excel or LibreOffice Calc.
If you do not have the results or were unable to run the DGE step, you can copy these two here which will be required for functional annotation (optional).
cp /sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/dge_results.txt .
cp /sw/share/compstore/courses/ngsintro/rnaseq/main/5_dge/dge_data.Rdata .